I Tumori del Sistema Nervoso Centrale
-
GASTROEPATO
-
Elenco
articoli di neurologia
-
I tumori del
sistema nervoso centrale
Si tratta di tessuti patologici, appunti neoplasie o tumori, che si
sviluppano dentro l'encefalo. Queste neoplasie possono essere più o meno
aggredibili, nel senso che il neurochirurgo, avvalendosi di tecniche differenti,
può riuscire ad eliminare le lesioni e ad avere un buon successo nella cura
stessa del tumore. Questo accade per il fatto che i tumori encefalici non danno
metastasi a distanza perchè il cervello è privo di linfatici, cioè di vasi di
drenaggio della linfa e di conseguenza di vasi che trasportano cellule tumorali
disseminandole altrove. Questa evenienza non capita, per esempio, nel caso di
tumori dei tessuti solidi. In genere un paziente con neoplasia nel suo cervello
si accorge di questa per una cefalea insistente ed insopportabile, perchè il suo
sguardo devia e gli occhi appaiono fuori asse, oppure perché avverte un
improvviso malessere, per es. con fatti convulsivi,
fino ad arrivare a sonnolenza o coma o a perdita di forza. A questo punto viene
effettuata un'indagine al suo cervello, in genere una tc encefalo, che documenta
la lesione, sia essa francamente encefalica o a partenza da strutture della
scatola cranica, quali le meningi, che accrescendosi ha determinato compressione
sull'encefalo stesso. In questo caso, di dice tecnicamente che la lesione è una
LOS, cioè una lesione occupante spazio.
I tumori a sede cerebrale differiscono da quelli di altre sedi per alcune
ragioni:
a) non danno metastasi
b) parimenti quelli benigni e cosi pure i maligni danno preoccupazione per
via dell'accrescimento che può determinare fenomeni compressivi pericolosi
c) l'inquadramento classificativo di questi
tumori (istologico, grading, frequenza e distribuzione per età e sede e cosi
via) è stata rivista nel 2016.
I tumori del sistema nervoso centrale possono avere una sede:
- Intracranica
- Spinale
Gli effetti che una neoplasia del SNC produce dipendono dalla sede in cui si
localizza, con segni "centrali" o "periferici". Quelli a sede intracranica danno sintomi differenti rispetto a quelli
localizzati nel midollo spinale. In quest'ultimo caso, infatti, potrò avere dei
sintomi periferici, per la compressione esercitata dalla massa che cresce e che dipendono dalla sede della neoplasia (parestesie,
alterazioni motorie).
Nella diagnostica delle neoplasie del SNC una delle prime cose importanti è
l'esame obiettivo neurologico accurato perché questo consente più o meno di
stabilire la sede della neoplasia.
I sintomi, tuttavia, non sono sempre specifici. Le lesioni intracraniche danno
sintomi
comuni ad altre patologie. I pazienti
con un'emorragia subaracnoidea avranno una sintomatologia di tipo acuto, quelli
con una neoplasia, soprattutto se questa è a lento accrescimento, riferiranno
sintomi che si protraggono da mesi o da anni. è importante quindi una corretta
anamnesi. Uno dei sintomi più frequenti è la cefalea, ovviamente per i tumori
intracranici. Altri sintomi sono: il vomito, modificazioni della personalità,
deficit motori, crisi epilettiche (in genere nei tumori localizzati a livello
corticale perché c'è un'irritazione della corteccia corticale).
In tutte le neoplasie che riguardano il SNC una caratteristica è l'aumento della
pressione endocranica. La scatola cranica è un compartimento chiuso
dove sono allocati: il parenchima cerebrale, il sangue e il liquor cefalorachidiano. Al suo interno c'è una determinata pressione ben precisa in
quanto queste componenti sono in equilibrio tra di loro. Se uno di questi
componenti aumenta di volume aumenta la pressione endocranica. Se ho un tumore
aumenta la quota di parenchima cerebrale, se ho un'emorragia aumenta la quota
ematica, se ho un idrocefalo aumenta il liquor (legge di Monro-Kelly).
L'ipertensione endocranica non è specifica delle neoplasie ma la posso trovare
in una serie di processi. Quello che si verifica è che il parenchima cerebrale
potrà farsi spazio attraverso il forame occipitale. La compressione dei vasi da
parte del parenchima spostato è responsabile del presentarsi della cefalea, del
vomito e dell'edema della papilla ottica (triade sintomatologica caratterista di
ipertensione endocranica e quindi presente anche nelle meningiti, negli
ascessi).
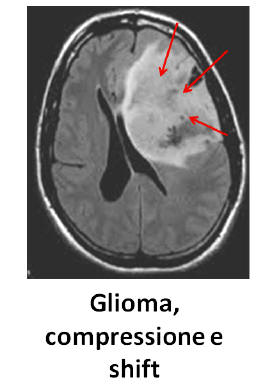 La
malignità dei tumori cerebrali è diversa da quelli dei tumori di altre sedi.
Nei tumori cerebrali non è importante considerare la loro benignità biologica, ma il fatto che una neoplasia, se non resecata, può comunque portare a morte il paziente perché
occupa spazio, produce,
cioè, degli spostamenti del parenchima
che possono determinare un'erniazione delle tonsille cerebellari verso il basso
che possono essere responsabile di compressione a livello del tronco
dell'encefalo. Poiché qui vi sono i centri cardiaci e respiratori bulbari il
paziente va in contro a morte. Quindi anche i processi benigni possono portare a
morte il paziente per queste dislocazione. La rapidità di questo dipende da dove
è localizzato il processo neoplastico. Un tumore cerebellare e un tumore
frontale non hanno la stessa emergenza chirurgica in quanto il primo è più
propenso all'emiazione delle tonsille cerebellari e, dunque, più pericoloso per
la compressione sui nuclei respiratori e cardioacceleratori. Quindi possiamo dire che in
generale i processi localizzati in fossa cranica posteriore rappresentano
emergenza chirurgica maggiore rispetto a quelli a sede frontale. Quindi se io ho
un paziente con una metastasi cerebellare, anche se l'intervento non è curativo,
questa va resecata perché altrimenti il paziente muore. Quindi qualsiasi tumore
cerebellare è potenzialmente maligno. Se pensiamo ad un tumore spinale,
localizzandosi in alcune sedi, può dare una paralisi completa quindi pur non
portando a morte il paziente ne compromette parimenti gravemente la vita. I tumori del
tronco cerebrale, pur essendo benigni possono portare a morte il paziente.
La malignità dei tumori cerebrali dipende anche dalla possibilità che
hanno questi di essere aggrediti chirurgicamente. I tumori della base
cranica sono difficilmente asportabili mentre quelli del tronco cerebrale non
sono asportabili perché l'intervento chirurgico potrebbe lesionare una delle
strutture vitali qui presenti (si fa unicamente una biopsia per determinare la
possibilità di fare un trattamento radio-chemioterapico). Anche tumori
spinali non sempre sono totalmente asportabili. Altre volte, anche se il tumore
ha una sede aggredibile, potrebbe essere localizzato vicino ad un vaso, come la
carotide, per cui non si può asportare in maniera completa senza essere sicuro
di danneggiare la carotide. Anche i tumori molto estesi non possono essere
sottoposti a chirurgia.
La
malignità dei tumori cerebrali è diversa da quelli dei tumori di altre sedi.
Nei tumori cerebrali non è importante considerare la loro benignità biologica, ma il fatto che una neoplasia, se non resecata, può comunque portare a morte il paziente perché
occupa spazio, produce,
cioè, degli spostamenti del parenchima
che possono determinare un'erniazione delle tonsille cerebellari verso il basso
che possono essere responsabile di compressione a livello del tronco
dell'encefalo. Poiché qui vi sono i centri cardiaci e respiratori bulbari il
paziente va in contro a morte. Quindi anche i processi benigni possono portare a
morte il paziente per queste dislocazione. La rapidità di questo dipende da dove
è localizzato il processo neoplastico. Un tumore cerebellare e un tumore
frontale non hanno la stessa emergenza chirurgica in quanto il primo è più
propenso all'emiazione delle tonsille cerebellari e, dunque, più pericoloso per
la compressione sui nuclei respiratori e cardioacceleratori. Quindi possiamo dire che in
generale i processi localizzati in fossa cranica posteriore rappresentano
emergenza chirurgica maggiore rispetto a quelli a sede frontale. Quindi se io ho
un paziente con una metastasi cerebellare, anche se l'intervento non è curativo,
questa va resecata perché altrimenti il paziente muore. Quindi qualsiasi tumore
cerebellare è potenzialmente maligno. Se pensiamo ad un tumore spinale,
localizzandosi in alcune sedi, può dare una paralisi completa quindi pur non
portando a morte il paziente ne compromette parimenti gravemente la vita. I tumori del
tronco cerebrale, pur essendo benigni possono portare a morte il paziente.
La malignità dei tumori cerebrali dipende anche dalla possibilità che
hanno questi di essere aggrediti chirurgicamente. I tumori della base
cranica sono difficilmente asportabili mentre quelli del tronco cerebrale non
sono asportabili perché l'intervento chirurgico potrebbe lesionare una delle
strutture vitali qui presenti (si fa unicamente una biopsia per determinare la
possibilità di fare un trattamento radio-chemioterapico). Anche tumori
spinali non sempre sono totalmente asportabili. Altre volte, anche se il tumore
ha una sede aggredibile, potrebbe essere localizzato vicino ad un vaso, come la
carotide, per cui non si può asportare in maniera completa senza essere sicuro
di danneggiare la carotide. Anche i tumori molto estesi non possono essere
sottoposti a chirurgia.
Stessa cosa se i tumori sono multipli.
In base alla sede, distinguiamo i tumori intracranici in tre categorie:
1. sopra-tentoriali o della fossa cranica anteriore
2. infra-tentoriali o della fossa cranica media
3. sotto-tentoriali o della fossa cranica posteriore
Quando io apro la scatola cranica, asporto il cervello sulla superficie
interna delle ossa del tavolato cranico vedo la dura madre mentre le
leptomeningi rimangono adese al cervello. La dura madre fa dei sepimenti che
sono:
a. la falce cerebrale che separa i due emisferi cerebrali
b. il tentorio che separa il lobo occipitale dal cervelletto.
La falce si trova quindi sulla linea sagittale, mentre trasversalmente ho il
tentorio. Il tentorio possiede un forame detto forame del tentorio. Se io
ho una neoplasia che si trova a livello del lobo frontale sinistro questa
spingerà il parenchima cerebrale inizialmente al di sotto della falce producendo
uno spostamento detto erniazione sub-falcina. Guardando un'immagine radiologica
si vede il parenchima cerebrale deformato con i ventricoli deformati e uno
spostamento al di sotto della falce. Dopo di ciò il parenchima cerebrale
spingendo verso il basso incontra il tentorio potendo passare attraverso il
forame del tentorio. Quindi in questo caso ho un'erniazione trans-tentoriale.
Alla fine continuerà a spingere producendo lo spostamento del cervelletto
all'interno del forame occipitale con erniazione tonsillare. Quindi se io ho una
neoplasia del lobo frontale prima da l'erniazione subfalcina, poi
trans-tentoriale e poi tonsillare. Se io ho una lesione a livello cerebellare
darà direttamente l'erniazione tonsillare. La rapidità con cui si può avere il
decesso del paziente è diversa nelle due condizioni. L'erniazione del tentorio
può determinare la compressione del mesencefalo.
L'eziologia dei tumori cerebrali è per gran parte sconosciuta. Esistono delle
sindromi ereditarie che si associano allo sviluppo di tumori del SNC:
1. Neurofibromatosi 1 e 2, sono associate allo sviluppo sia di tumori delle
meningi che sono i meningiomi, sia di tumori che originano dalle cellule di
Shwann che si chiamano Shwannomi.
2. Sclerosi tuberosa
3. Sindrome di Turcot: porta allo sviluppo di medullo-blastoma e di carcinoma
del colon-retto.
4. Sindrome di Von Hippel Lindau: porta allo sviluppo di tumori cerebellari
detti emangioblastomi, carcinoma renale e emangiomi retinici.
5. Sindrome di Li Fraumeni
6. Sindrome di Cowden
Uno dei fattori scatenanti sono le radiazioni, per cui pazienti che subiscono
radioterapia sull'encefalo hanno una maggiore possibilità di sviluppare delle
altre neoplasie dopo il trattamento.
Epidemiologia
I tumori del SNC sono delle neoplasie relativamente rare che costituiscono circa
il 2% di tutte le neoplasie dell'adulto e il 20% delle neoplasie pediatriche.
Rispetto al passato questi dati sono in aumento perché prima erano
sottodiagnosticati o misdiagnosticati per patologie psichiatriche
I tumori del SNC non hanno la stadiazione TNM.
La T negli organi parenchimatosi corrisponde alle dimensioni del tumore ma in
questa sede non sono importanti tanto le dimensioni quanto il tipo di tumore,
l'istologia e la sede per cui la dimensione non ha rilevanza prognostica.
Non avendo linfatici l'N non risulta applicabile (niente linfonodi e
metastasi nei linfonodi)
Le metastasi a distanza sono rarissime perché le metastasi sono
pressocchè impossibili, se non , raramente col liquor.
Attualmente il fattore più importante per questi tumori è la radicalità
chirurgica.
Classificazione delle neoplasie del SNC secondo OMS 2016
Per tutte le Neoplasie esiste una classificazione fatta dall'OMS la quale
riunisce degli esperti e classifica questi tumori. Le classificazioni vengono
illustrate all'interno dei cosiddetti "bluebooks". Sono classificazioni che
tendono a variare con l'aumento delle conoscenze. Nel caso del SNC l'ultima
classificazione è quella del 2016 la quale divide i tumori in diverse categorie
e ci dà delle informazioni per il loro grading.
Nelle neoplasie del SNC il grading non si basa soltanto sul grado di
differenziazione (come per gli altri distretti) ma incorpora altri elementi. è
un sistema articolato su 4 gradi diversi di malignità ed è basato sul decorso
clinico. Vedremo che ogni neoplasia ha abbinato un grado istologico. Per esempio
il glioblastoma che è una neoplasia che deriva dagli astrociti è per definizione
un grado 4. Questo perché si è visto qual è il comportamento clinico nel tempo e
siccome il glioblastoma porta in genere a decesso i pazienti entro 18 mesi è
considerato come grado 4.
Il grado è molto importante anche per la terapia
perché i tumori di grado 1 se fanno un'asportazione chirurgica radicale non
devono fare altre terapie, tumori di altro grado fanno anche altre terapie come
la chemio o la radioterapia
1. Il grading dei tumori cerebrali ha tra i suoi parametri il pattern di
crescita.
Sapete che possiamo avere tumori a crescita espansiva o tumori a crescita infiltrativa. Un esempio di tumore espansivo fuori dal SNC sono i
leiomiomi. Nel
SNC i tumori che hanno crescita espansiva vengono considerati tumori di grado 1,
quelli a crescita infiltrativa possono essere di grado 2, 3 o 4. La differenza
sta nel fatto che i tumori infiltrativi hanno maggiori difficoltà ad essere
asportati in modo radicale. Questo anche perché i margini di resezione non
possono essere molto ampi. Quindi le recidive sono più probabili. Dalla capacità
proliferativa di queste cellule dipende il timing della recidiva. Sta qui la
differenza tra i tumori di grado 2, 3 e 4. Tutti proliferano ma lo fanno con
velocità diversa.
2. Altro parametro per il grading è la capacità proliferativa. La proliferazione
è valutata in base al numero di mitosi e in base alla presenza di necrosi.
Es. in un tumore di grado due mi aspetto: una proliferazione bassa, assenza di
necrosi. In un tumore 3 o 4 abbiamo mitosi piuu numerose e può esserci la
comparsa di necrosi.
Per quanto riguarda le classificazioni dell'OMS, queste si sono basate sulla
morfologia delle cellule.
Nel 2016 sono cambiate un po' di cose. Sono tumori che originano dalle cellule
gliali e possono essere, secondo la classificazione 2007:
- Astrocitomi (dagli astrociti):
1. diffusi, ovvero a crescita infiltrativa.
a. di grado 2
b. di grado 3 detto anche Anaplastico
c. di grado 4 detto anche Glioblastoma che si suddivide in:
• Primario: grado 4 de novo. Caratteristico dei soggetti con età superiore a 50
anni
• Secondario: deriva da astrocitomi di grado più basso. Caratteristico dei
giovani.
2. Localizzati, ovvero a crescita espansiva o di grado 1
- Oligodendrogliomi (degli oligodendrociti) Possono essere:
1. Grado 2
2. Grado 3
Non esiste un oligodendroglioma di grado 1 perché sono tumori infiltranti. Non
esiste nemmeno un grado 4 perché la prognosi degli oligodendrogliomi è migliore
di quella dei glioblastomi. Nel 2007 erano indicati dal punto di vista
morfologico come tumori caratterizzati da cellule rotondeggianti con alone
chiaro perinucleare che gli conferiva un aspetto a uovo fritto
- Oligoastrocitomi. Il tumore ci indica che questi tumori hanno delle
caratteristiche istologiche intermedie tra i tumori oligodendrogliali e i tumori
astrocitari. Gli oligoastrocitomi potevano essere di grado due o di grado 3.
 - Ependimomi (tumori che si ritiene derivino dall'ependima) Caratteristiche
molecolari dei gliomi
- Ependimomi (tumori che si ritiene derivino dall'ependima) Caratteristiche
molecolari dei gliomi
Ma cosa è cambiato nella classficazione del 2016 da quella del 2007? Vengono
inseriti i dati molecolari. Non basta infatti l'osservazione istologica ma è
necessario sapere il tipo molecolare di quel tumore perche alcuni tumori sono
definiti dalle loro caratteristiche molecolari.
Uno dei lavori scientifici che ha indirizzato in questo senso è un lavoro de
2008 che ha evidenziato per la prima volta nei glioblastomi delle mutazioni
ricorrenti di un gene detto Isocitrato Deidrogenasi IDH. è un lavoro del
tutto
casuale perchè questi autori studiavano il profilo genetico di alcuni tumori e i
glioblastomi si prestavano a questo scopo perché i pazienti muoiono nel giro di
12-18 mesi quindi non c'era bisogno di avere il consenso.
L'isocitrato-deidrogenasi esiste in due isoforme: IDH1 e IDH2. Catalizzano la
conversione dell'isocitrato ad alfachetoglutarato. Quando questi enzimi sono
mutati portano alla formazione in elevate quantità di 2-idrossi-glutarato che
induce la metilazione del DNA che induce la tumorigenesi. Quindi la mutazione
dei geni IDH1 e IDH2 induce la formazione di tumori non soltanto nel SNC ma
anche nel carcinoma correttale. In questa pubblicazione sono state analizzate
delle mutazioni di IDH1 e IDH2 in una serie di tumori del SNC e si è visto che
esistono delle mutazioni ricorrenti in determinate posizioni dei due geni di cui
circa il 90% si localizza in una posizione specifica del gene IDH1 e queste
mutazioni sono particolarmente frequenti negli astrocitomi di grado 2 e 3 e nel
glioblastoma secondario ovvero quello che deriva da un astrocitoma di grado più
basso. Queste mutazioni non sono presenti nel glioblastoma primario né in quelli
pediatrici né in quelli di grado 1. Sono assenti anche negli ependimomi. Sono
presenti negli oligodendrogliomi. Questo è importante perché ha rilevanza
prognostica. Andando a prendere astrocitomi di grado 3 e grado 4 si è visto che,
nonostante si pensasse che il grado tre desse una prognosi migliore, l'astrocitoma
di grado 4 con mutazione di IDH va meglio rispetto ad un astrocitoma di grado 3
senza la mutazione. Quindi la mutazione ha un significato prognostico positivo.
Lo stato di mutazione è quindi più importante rispetto al grading istologico.
Questo ha rivoluzionato tutto il capitolo dei gliomi perché questo ha delle
conseguenze anche sul trattamento e sulla prognosi. La terapia dei gliomi IDH
wildtype, quindi non mutata, è più aggressiva di quella invece per i tumori con
mutazione. Questi dati sono stati quindi incorporati nella classificazione. Le
mutazioni per IDH si valutano all'immunoistochimica (con anticorpi marcati
diretti verso la mutazione di IDH più frequente ovvero R132H). Se l'immunoistochimica
è negativa quindi l'anticorpo non reagisce questo non vuol dire che non ci siano
mutazioni in quanto potrebbero esserci delle mutazioni diverse da quelle
riconosciute dall'anticorpo oppure una mutazione del gene IDH2. Quindi in questo
caso per rilevare la presenza di mutazioni ho bisogno del sequenziamento genico
per i geni IDH1 e IDH2 che mi dice se ci sono mutazioni o no.
Altra caratteristica molecolare dei tumori gliali e in particolare dell'oligodendroglioma
è la presenza della codelezione 1p\19q. Questa
codelezione è specifica dell'oligodendroglioma ed è importante sapere se c'è o
meno perché i tumori con questa codelezione hanno una maggiore sensibilità alla
chemioterapia e hanno anche una prognosi migliore.
Altra mutazione che si è trovata nei gliomi a parte IDH1, è quella di un gene
detto ATRX (alpha-talassemia-mental-retardation-syndrome-x) la cui mutazione la
osserviamo negli astrocitomi ed è strettamente associata alla mutazione di IDH1
ed è mutualmente esclusiva con 1p\19q.
Quindi:
a. i tumori che hanno la mutazione ATRX in genere sono astrocitomi
b. quelli con la mutazione 1p\19q sono oligodendrogliomi.
Questo ha portato un approccio differente nella diagnosi dei gliomi. Quindi gli
astrocitomi non vengono più classificati in base all'aspetto istologico ma in
base a quello molecolare. L'astrocitoma di grado 2 e 3 ha: mutazione di IDH,
mutazione di ATRX e mutazione di P53. L'oligodendroglioma ha una mutazione di
IDH e codeezione 1p\19q.
Per quanto riguarda gli oligoastrocitomi tutt'ora presenti nella classificazione
sono una diagnosi scoraggiata. Precedentemente veniva fatta più per incapacità
del patologo a distinguere astrocitoma e oligodendroglioma oggi cosa fattibile
tramite i dati molecolari. Quindi è una diagnosi che facciamo se non possiamo
avere il dato molecolare.
Quindi in base all'avanzamento delle conoscenze, i tumori astrocitari e
oligodendrogliali dal 2016 non vengono più classificati sul dato morfologico ma
anche sulla base dei dati molecolari.
Rispetto al 2007 i principali cambiamenti sono nella categoria dei tumori
astrocitari e oligodendrogliali e nella categoria dei tumori embrionari, quelli
del bambino. Quindi in base alla nuova classificazione abbiamo:
 è un grado 1 ed è una neoplasia
tipicamente del bambino o dell'adolescente che insorge a livello troncoencefalico o del cervelletto o a livello del talamo. La prognosi è
eccellente perché se viene asportato in maniera radicale a 25 sopravvivono più
del 95% dei pz. è chiaro che nella localizzazione troncoencefalica la prognosi
non sarà cosi buona perché sarà . difficile asportarlo. Non è un tumore
rapidamente fatale. Si presenta tipicamente con un nodulo all'interno di una
cisti. è un tumore con crescita espansiva. Si chiama pilocitico perché è
costituito da astrociti
è un grado 1 ed è una neoplasia
tipicamente del bambino o dell'adolescente che insorge a livello troncoencefalico o del cervelletto o a livello del talamo. La prognosi è
eccellente perché se viene asportato in maniera radicale a 25 sopravvivono più
del 95% dei pz. è chiaro che nella localizzazione troncoencefalica la prognosi
non sarà cosi buona perché sarà . difficile asportarlo. Non è un tumore
rapidamente fatale. Si presenta tipicamente con un nodulo all'interno di una
cisti. è un tumore con crescita espansiva. Si chiama pilocitico perché è
costituito da astrociti
con dei prolungamenti piloidi simili a capelli. è caratterizzato da aree
cistiche e da aree solide che sono quelle che abbiamo visto che producono il
liquido.
L'astrocitoma diffuso che è un tumore di grado 2, in quanto infiltrativo, ad
origine dagli astrociti che in genere colpisce l'adulto, può insorgere in tutto
il nevrasse e può avere una progressione verso forme di astrocitoma di grado più
elevato se non viene operato o se recidiva. Dal punto di vista macroscopico si
manifesta come un tumore a crescita infiltrativa perché neoplasia e parenchima
sano non sono facilmente distinguibili. In base alla nuova classificazione
esistono tre tipi di astrocitoma diffuso.
a. IDH mutato
b. IDH Wild-Type
c. NAS non altrimenti specificato, diagnosi che si fa quando non è possibile
fare i test molecolari
Questa classificazione è importante perché l'astrocitoma diffuso IDH mutato
verosimilmente andrà bene e se l'asportazione è macroscopicamente radicale non
fa altre terapie. La forma Wild-Type ha una prognosi peggiore quindi se non
viene asportato tutto deve fare radio e chemioterapia, quindi anche se è un
tumore di grado due viene trattato come un tumore d grado 4. La mutazione del
gene IDH ha una rilevanza sia per la prognosi che per la terapia. Dal punto di
vista istologico l'astrocitoma diffuso è una neoplasia a bassa densità
cellulare, non ci sono figure mitotiche. Dal punto di vista istologico di un
astrocitoma idh mutato o idh wildtype è lo stesso. Si deve fare quindi
un'indagine immunoistochimica con anticorpi diretti contro la proteina mutata
con mutazione R132H. Negli astrocitomi diffusi di grado due con mutazione IDH
c'è anche la mutazione del gene ATRX e la mutazione di p53.
Anche questa ovviamente una
neoplasia a differenziazione astrocitaria, in genere dell adulto le cui recidive
sono più frequenti rispetto al grado due in quanto prolifera più rapidamente.
Anche per quuesto tipo di astrocitoma posso avere la forma IDH mutata la forma
IDH wildtype e la forma NAS. L'aspetto istologico rispetto al grado due è
diverso perché la cellularità è maggiore e ho la presenza di atipie e mitosi.
Una cosa importante sul grading degli astrocitomi di grado 2 e grado 3 è
emersa con un lavoro pubblicato nel 2015. Gli astrocitomi erano suddivisi e sono
ancora suddivisi in grado due e grado tre perché la sopravvivenza di quelli di
grado tre era peggiore di quella dei grado due. Si è visto però che se io
considero soltanto gli astrocitomi di grado tre e di grado due che hanno la
mutazione di IDH non c'è più nessuna differenza prognostica. Quindi questo
indica che il grading è meno importante della mutazione. Se io ho un tumore con
mutazione IDH che sia grado due o tre si comporta allo stesso modo.
L'astrocitoma di grado 4 si chiama anche glioblastoma ed è una
neoplasia estremamente aggressiva e può essere primaria e insorgere direttamente
come grado 4 oppure secondaria se si evolve da un grado due o tre. L'aspetto
macroscopico è caratterizzato da necrosi e aree di emorragia che in passato gli
davano anche il nome di glioblastoma multiforme. Anche per il glioblastoma si
possono avere le tre forme: IDH mutato, IDH wildtype, NAS.
In genere il glioblastoma IDH mutato lo troviamo soprattutto in persone al d
sotto dei 50 anni di età, quello wildtype lo troviamo in persone con più di 50
anni di età. La prognosi è diversa perché la mutazione da una prognosi migliore.
Da un punto di vista istologico si distingue per una densità cellulare elevata,
presenza di proliferazione vascolare e di mitosi più frequenti del grado tre. è
presente anche necrosi a palizzata, al centro ho la necrosi e le cellule si
dispongono a formare delle palizzate intorno alla necrosi. I glioblastomi
possono a volte presentare alterazioni di un gene che è quello della
06-metil-guanin-DNAmetiltransfersi (MGMT). Quando tale gene ha la metilazione
del promotore non esprime la proteina che ha il compito fisiologico di riparare
i danni da agenti alchilanti. Quindi se ho la metilazione del promotore e non ho
più l'enzima il tumore risponderà di più alla terapia per cui nel glioblastoma
viene fatta l'analisi del gene MGMT per capire quanto il tumore risponderà alla
terapia. I glioblastomi pediatrici differiscono un po' dai tumori dell'adulto.
Un lavoro del 2012 ha effettuato l'analisi mutazionale di 48 glioblastomi
pediatrici che sono glioblastomi IDH wildtype. Hanno invece un'altra mutazione
che è la mutazione del gene H3F3A che codifica per una proteina istonica. Si
possono avere 2 tipi di mutazioni che sono: -K27M -G34B.
Questi due tipi di mutazioni le troviamo in tumori che si localizzano in sedi
diverse. La mutazione K27M è presente nei glioblastomi pediatrici che si trovano lungo la linea mediana, per esempio del talamo, del tronco
dell'encefalo. I glìoblastomi che hanno la mutazione del gene G34B si
localizzano negli emisferi cerebrali quindi la distribuzione delle due mutazioni
è differente per sede. Ma la cosa più interessante è che si è visto che i tumori
della linea mediana con la mutazione K27M vanno particolarmente male perché sono
molto aggressivi pur non essendo istologicamente aggressivi, lo posso avere un
tumore che istologicamente ha le caratteristiche di astrocitoma diffuso di grado
2 ma se ha la mutazione k27m si comporta come un glioblastoma. Anche in questo
caso la mutazione è più importante dell'aspetto istologico. Per questo è stata
introdotta una nuova entità tra i tumori astrocitari che non si trova nella
classificazione del 2007 e che si chiama "diffused midline glioma H3f3a, k27m
mutato". Questa entità nella classificazione 2016, a prescindere dall'aspetto
istologico viene considerata come grado 4 perché ha una prognosi cattiva. Se ho
un tumore della linea mediana devo quindi valutare se ho questa mutazione.
Questa mutazione si vede con un anticorpo all'immunoistochimica.
Tumori che
originano dagli oligodendrociti si chiamano oligodendrogliomi. Posso averne due
tipi: grado 2 e grado 3. Sono tumori piuttosto rari che possono insorgere in
qualsiasi parte del SNC sono tumori dell'adulto e sono sensibili alla chemioterpia. Questo è l'aspetto istologico, simile agli oligodendrociti
normali, ho delle cellule con un nucleo rotondeggiante e un alone chiaro
perinucleare. Queste cellule neoplastiche tendono a infiltrare la corteccia
cerebrale e a disporsi intorno ai neuroni dando delle immagini di satellitosi
perineuronale. Oligodendroglioma non viene più definito dall'aspetto istologico
ma è definito dalla codelezione 1p\19q. Quindi per essere sicuro di essere di
fronte ad un oligodendroglioma devo valutare la presenza di questa codelezione.
Se non sono in grado di fare i test molecolari lo definisco come NAS. Più
dell'aspetto istologico è la codelezione a dare la sensibilità alla
chemioterapia. LA codelezione si valuta col sequenziamento o con la FISH (ibridizzazione
in situ con sonde fluorescenti). Nella nuova classificazione l'oligodendroglioma
viene quindi definito come tumore con mutazione di IDH e codelezione di 1p\19q.
L'oligodendroglioma anaplastico, che è il grado 3, rispetto al grado due ha un
aumento e posso osservare la presenza di necrosi e di proliferazione vascolare.
Non ho un grado 4 di oligodendroglioma perche il genere va prognosticamente
meglio e non ho un grado di malignità sovrapponibile a quello del glioblastoma.
Anche per l'oligodendroglioma anaplastico ho mutazione di IDH e codelezìone di 1p\19q.
Se non posso eseguire i test lo diagnostico come NAS.
Rimane nella classificazione del 2016 l'entità oligoastrocitoma, tumore con
morfologia intermedia. Se io mi trovo in un'anatomia patologica non in grado di
valutare la mutazione di IDH e la codelezìone 1p\19q e ho un tumore con una
morfologia ambigua tra astrocitoma e oligodendroglioma posso utilizzare questa
diagnosi. è NAS perché io posso fare questa diagnosi solo se non ho i test
molecolari. Perché se ho i test molecolari mi troverò di fronte a un tumore che
ha la codelezìone 1p\19q e quindi è un oligodendroglioma oppure di fronte a un
tumore che ha solo la mutazione di IDH ed è unastrocitoma. Ci sono però rari
casi di tumori con doppio genotipo che hanno una popolazione di cellule con la
codelezìone e una popolazione con la mutazione di IDH allora in questi rari casi
posso parlare di oligoastrocitoma perche sono neoplasie miste anche da un punto
divista genetico.
Sono tumori che originano dalle cellule ependimali.
Ne
conosciamo diversi tipi:
1. sub-ependimoma che è un tumore di grado 1
2. ependimoma mixopapillare anch'esso di grado 1
3. ependimoma RELA fusion positivo grado 2
4. ependimoma anaplastico grado 3
Anche per l'ependimoma non esiste il grado 4 perché non c'è una neoplasia cosi
aggressiva come il glioblastoma. L'ependimoma di grado 2 lo ritroviamo sia nel
bambino che nell'adulto. In genere nel bambino la localizzazione è a livello del
4 ventricolo mentre nell'adulto la localizzazione più frequente è a livello
spinale. Macroscopicamente si presenta con un aspetto violaceo che indica una
spiccata vascolarizzazione. Istologicamente forma rosette e canali ependimali
mimando la loro azione fisiologica. Un'altra caratteristica sono le
pseudorosette vascolari, in cui le cellule neoplastiche si dispondono attorno
alle strutture vascolari.
Nell'ependimoma anaplastico di grado 3 rispetto al grado 2 ho un aumento deila
densità cellulare, la presenza di mitosi, la presenza di necrosi e di
proliferazione vascolare.
Una novità è l'ependimoma RELA Fusion positivo. Anche questa è un'entità basata
su studi molecolari. è un ependimoma che ha una fusione tra due geni: il gene
RELA e un altro gene presente sul cromosoma 11. Questo ependimoma colpisce sia adulti che bambini e la
caratteristica che lo distingue è che è solo sopratentoriale. è importante
distinguerla perché i dati ci dicono che gli ependimomi sopratentoriali con
questa alterazione genetica hanno una prognosi peggiore rispetto agli altri
ependimomi. Ancora non c'è una classificazione definitiva degli ependimomi ma si
è visto che quelli sopratentoriali hanno delle modificazioni genetiche, quelli
della fosse cranica posteriore ne hanno delle altre e quelli del midollo spinale
delle altre ancora, nonostante lo stesso aspetto istologico. Altro tipo di
ependimoma è l'ependimoma mixopapillare che è piuttosto raro, è un grado 1 e si
localizza a livello della cauda equina, è benigno e si asporta facilmente senza
dare recidive. Si chiama mixopapillare perche forma delle papille all'interno
delle quali c'è un materiale mixoìde.
Tra gli atri gliomi troviamo quelli dei plessi corioidei si trovano a livello
delle cavità ventricolari e producono il liquor cefalorachidiano. Sono tumori
epiteliali e la loro nomenclatura è uguale a quella dei tumori epiteliali. Posso
avere:
1. papilloma, tumore di grado 1 benigno.
2. papilloma atipico, tumore a malignità intermedia di grado 2
3. carcinoma dei plessi corioidei. Grado 3
Tra i tumori embrionari il più frequente è il medulloblastoma che è un
tumore di grado 4 per definizione. In genere è dell'età pediatrica e si
localizza a livello del cervelletto, nei bambini a livello del verme, nei rari
casi dell'adulto si localizza negli emisferi cerebellari. Ha una crescita molto
rapida e si diffonde attraverso il liquor cefalo-rachidiano che deve quindi
essere analizzato per vedere se ci sono cellule tumorali. Si deve fare anche un
RMN del midollo spinale per vedere se ci sono impianti che si sono diffusi
attraverso il LCR. Ha una densa cellularità, mitosi frequenti, posso trovare
necrosi e proliferazione vascolare. Spesso ha una differenziazione neuronale e
le cellule neoplastiche si possono disporre a formare delle rosette dette di
Homer-Wright. Avendo una differenziazione neuronale esprima markers neuronali
come la sinaptofisina. La valutazione di Ki 67 nel medullo blastoma arriva anche
al 90% è quindi un tumore ad alta attività proliferativa. Esistono diverse
varianti istologiche:
1. medulloblastoma classico
2. medulloblastoma desmoplastico-nodulare
3. medullo blastoma con estensiva nodularità
4. medulloblastoma a grandi cellule o anaplastico
Questi sottogruppi istologici hanno un'importanza istologica in quanto il
medulloblastoma di tipo nodulare è quello dalla prognosi migliore, quello a
grandi cellule o anaplastico ha la prognosi peggiore.
Nella nuova classificazione sono stati introdotti anche i tipi molecolari in
base ai quali il medullo blastoma viene suddiviso in 4 gruppi:
1. Gruppo con alterazioni del pathway di WNT ed è quello dalla prognosi
migliore.
2. Gruppo con alterazioni SHH (sonic hedge-hog)
3. Gruppo 3
4. Gruppo 4
Hanno prognosi e terapia differente. Il primo gruppo è quello che va meglio, i
gruppi 3 e 4 sono quelli che vanno peggio. Quindi è importante sia la
classificazione istologica che quella molecolare. Anche in questo caso se non
posso fare i test molecolari rimane la categoria NAS. La diagnosi non ha bisogno
di attrezzature particolari perche con l'utilizzo di 4 anticorpi che sono: GAB1,
betacatenina, filamina e YAP.
Tumori dei nervi cranici e paraspinali
Il più diffuso è lo Shwannoma, è un tumore di grado 1 a crescita espansiva,
aderisce alle radici nervose, si riesce facilmente ad eradicare in toto e la sua
più frequente localizzazione a livello del SNC è a livello della branca acustica
dell'ottavo nervo cranico. Si manifesta con alterazioni dell'udito e si trova a
livello dell'angolo ponto-cerebellare. è una neoplasia di colore giallastro,
circoscritta e istologicamente ha un'alternanza di aree lasse e compatte. Le
cellule neoplastiche si possono disporre a formare delle palizzate nucleari che
vengono dette corpi di Verocay.
Tumori a cellule germinali
Del tutto simili ai tumori del testicolo e dell'ovaio, sono in genere tumori
dell'infanzia e si localizzano sulla linea mediana a livello della regione
sellare e della regione pineale. Il più frequente è il germinoma uguale al
disgerminoma e al seminoma.
Tumori che originano dalle meningi
Possono originai dalle cellule meningoteliali e si chiamano meningiomi oppure
dai tessuti mesenchimali. I meningiomi originano dalle cellule meningoteliaii
presenti a livello dell'aracnoide e in particolare a livello delle granulazioni
del pacchioni. Potrò avere sia meningiomi intracranici che spinali. In genere
sono tumori benigni. Quando sono maligni possono infiltrare l'osso, il
parenchima cerebrale. Una delle caratteristiche istologiche è la presenza di
corpi psammomatosi che sono concrezioni calcaree (anche nel carcinoma papillare
della tiroide e dell'ovaio). Una delle caratteristiche dei meningiomi è quella
di formare delle strutture vortcoidi in cui si dispongono le cellule a bulbo di
cipolla emulando le cellule meningoteliali normali che troviamo nelle
granulazioni del Pacchioni. Sono neoplasie degli adulti, soprattutto del sesso
femminile perché si pensa che abbiano una crescita correlata alla presenza di
ormoni sessuali tant'è che spesso questi tumori si sviluppano durante la
gravidanza. Secondo alcuni ci sarebbe anche un'associazione con l'uso di
contraccetivi. Altro fattore di rischio è la presenza di neurofibromatosi. I
pazienti affetti da questa patologia tendono infatti a formare meningiomi
multipli per la mutazione del gene NF2. Sono lesioni dure rotondeggianti che
aderiscono alla dura ma che sono facilmente enucleabili. Nella maggiorparte dei
casi hanno una crescita di tipo espansivo. Possono improntare il parenchima
cerebrale senza infiltrarlo. Ne esistono 15 diversi istotipi e 3 gradi di
malignità (1,2,3). Quelli di grado 1 sono l'80% e sono caratterizzati da un
basso indice mitotico (<4 mitosi per 10 campi ad alto ingrandimento) non
infiltrano il parenchima e hanno un lento accrescimento. Gli istotipi più
frequenti del grado 1 sono:
- Il meningioma meningoteliale, tende caratteristicamente a formare strutture
vorticoidi
- Il meningioma fibroblastico le cellule somigliano ai fibroblasti
- Meningioma transizionale, caratteristiche intermedie tra i primi due
Una minor quota di meningiomi sono di grado 2 (15-20%). Hanno un accrescimento
più rapido e tendono alla recidiva. Sono più frequenti nel sesso maschile. Dopo
l'asportazione dovrebbe andare alla radio sebbene non vi sia unanimità a
riguardo. Gli istotipi de grado due sono:
- Meningioma atipico: almeno 4 mitosi per 10 campi ad alto ingrandimento oppure
l'infiltrazione del parenchima cerebrale.
- Meningioma a cellule chiare
- Meningioma cordoide
Una piccola percentuale di meningiomi sono di grado 3, sono estremamente
aggressivi. Non ho il problema delle recidive perche sono tumori mortali.
Gli istotipi sono 3
- Meningioma anaplastico
- Meningioma rabdoide
- Meningioma papillare
Tumori della regione sellare
Sono i tumori della neuroipofisi e dell'asse ipotalamo ipofisario come il
craniofaringioma. è un tumore raro dall'aspetto cistico con un liquido denso
giallastro. Se ne distinguono due istotipi che sono:
- Craniofaringioma papillare, con mutazione del gene BRAF come il melanoma
- Craniofaringioma adamantinomatoso
Tumori metastatici
Tra i più frequenti. Da carcinoma del polmone, della mammella, rene e del tratto
gastroenterico. Col miglioramento delle tecniche si vedono metastasi che prima
non era possibile vedere.
indice di neurologia